
服務(wù)熱線:1362169548613621695486
服務(wù)熱線:1362169548613621695486
沒(méi)藥為橄欖科(Burevaceae)沒(méi)藥屬(Commiphora) 植物地丁樹(shù) Commiphora myrrha Egnl. 或哈地丁樹(shù) Commiphora molmol Egnl. 的 干 燥 樹(shù) 脂,分 為 天 然 沒(méi) 藥和膠質(zhì)沒(méi)藥,主產(chǎn)于索馬里與埃塞俄比亞 ,其質(zhì) 量標(biāo)準(zhǔn)收載于《中華人民共和國(guó)藥典》2015 年版一 部 。沒(méi)藥藥性辛、苦,平,歸心、肝、脾經(jīng),有特異的 香氣,在中國(guó)以及印度等地具有悠久的用藥歷史,具 有活血、消腫、止痛的功效,主治瘀血心腹諸痛、跌撲 傷痛等 。
安 捷 倫 公 司 Agilent 1260 型高效液相色譜儀,包括 G1311C 四元泵、在線脫氣機(jī)、G1315D DAD、G1329B 自 動(dòng) 進(jìn) 樣 器、LC1260 色 譜 工 作 站、 G1316A 柱溫箱;賽多利斯公司 Sartorius BT25S 電子 天平(十萬(wàn)分之一)、Sartorius BSA124S-CW 電子天 平(萬(wàn)分之一);昆山市超聲儀器公司 KQ-500DE 型 數(shù)控超聲波清洗器。
化合物Ⅰ、Ⅱ和Ⅲ的對(duì)照品均為本課題 組自制,通過(guò) 1H-NMR、13C-NMR、MS 等手段確證 了其結(jié)構(gòu),純度經(jīng) HPLC 法檢測(cè)均 >98%(面積歸一 法)。乙腈(Sigma 公司)為色譜純,水為 Milli-Q 超 純水,甲醇及乙醇(國(guó)藥集團(tuán)化學(xué)試劑有限公司)、磷 酸(西隴科學(xué)股份有限公司)為分析純。6 批沒(méi)藥藥 材均購(gòu)自于藥材市場(chǎng),由周國(guó)平主任藥師鑒定為地丁 樹(shù) Commiphora myrrha Engl. 的干燥樹(shù)脂。
精密稱取化合物Ⅰ、Ⅱ和Ⅲ 的對(duì)照品適量,加甲醇制得質(zhì)量濃度分別為 0.018 4、 0.029 0 和 0.025 8 mg·mL-1 的混合溶液,即得。
取沒(méi)藥藥材粉末(過(guò) 3 號(hào)篩)約 0.2 g,精密稱定,置具塞錐形瓶中,精密加入甲醇 25 mL,密塞,稱量,超聲(功率 500 W,頻率 40 kHz)處 理 0.5 h,放冷,再稱量,用甲醇補(bǔ)足減失的量,搖勻, 過(guò) 0.45 μm 微孔濾膜,即得。
采用資生堂 CAPCELL PAK C18 色譜柱(250 mm×4.6 mm,5 μm),柱溫 30 ℃,以乙腈(A)-0.1% 磷酸水溶 液(B)為流動(dòng)相,梯度洗脫(0~40 min,41%B;41~60 min,44%B),流速 1.0 mL·min-1,檢測(cè)波長(zhǎng) 210 nm,進(jìn)樣 量 10 μL;在上述色譜條件下,樣品色譜中與各對(duì)照品對(duì) 應(yīng)的吸收峰的理論板數(shù)均不低于 3 000,色譜圖見(jiàn)圖 1。
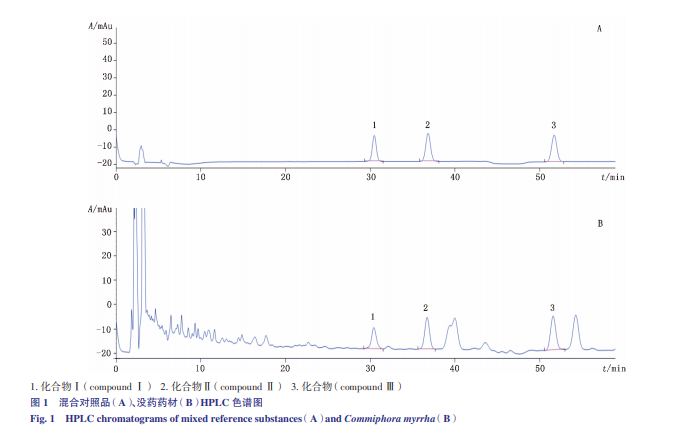
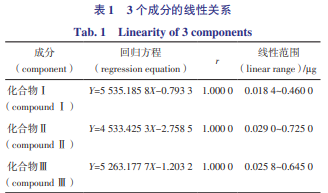
精密吸取“2.1”項(xiàng)下混合對(duì)照 品溶液 1、2、5、10、15、25,分別按“3”項(xiàng)下色譜條件 進(jìn)樣分析,以進(jìn)樣量 X(μg)為橫坐標(biāo),峰面積 Y 為縱 坐標(biāo),繪制標(biāo)準(zhǔn)曲線并進(jìn)行回歸計(jì)算,得 3 個(gè)成分的 線性回歸方程,結(jié)果見(jiàn)表 1。
精密吸取混合對(duì)照品溶液 5 μL, 按“3”項(xiàng)下色譜條件連續(xù)進(jìn)樣 6 次,依次測(cè)定峰面 積,結(jié)果化合物Ⅰ、Ⅱ和Ⅲ峰面積的 RSD(n=6)分別0.35%、0.17%、0.11%,表明儀器精密度良好。
精密吸取供試品溶液 10 μL,按 “3”項(xiàng)下色譜條件,于 0、1、2、4、8、12、24 h 分別進(jìn) 樣測(cè)定,結(jié)果化合物Ⅰ、Ⅱ和Ⅲ峰面積的 RSD(n=7) 分別為 0.62%、0.70%、0.69%,表 明 供 試 品 溶 液 在 24 h 內(nèi)穩(wěn)定。
取同一批沒(méi)藥藥材粉末(過(guò) 3 號(hào) 篩)6 份,各約 0.2 g,精密稱定,按“2.2”項(xiàng)下方法制 備供試品溶液,并按“3”項(xiàng)下色譜條件進(jìn)樣測(cè)定, 化合物Ⅰ、Ⅱ和Ⅲ的平均含量(n=6)分別為 0.791、 1.430、1.471 mg·g -1,RSD 分別為 0.91%、1.1%、 0.76%。
檢測(cè)波長(zhǎng)的選擇 應(yīng)用 DAD 檢測(cè)器在 190~400 nm 范圍內(nèi)對(duì)化合物Ⅰ、Ⅱ和Ⅲ進(jìn)行光譜掃描;結(jié)果 表明,在 210 nm 波長(zhǎng)附近 3 個(gè)成分吸光系數(shù)均最大, 干擾少且穩(wěn)定,故選擇 210 nm 作為檢測(cè)波長(zhǎng)。供試品溶液制備方法的考察 考察了回流提取 法和超聲提取法對(duì)化合物Ⅰ、Ⅱ和Ⅲ含量測(cè)定的影 響,結(jié)果顯示超聲提取法方便且能提取完全,故選擇 超聲提取法。提取溶劑考察了乙腈溶液、甲醇溶液、 乙醇溶液,結(jié)果顯示甲醇溶液提取時(shí)化合物Ⅰ、Ⅱ和 Ⅲ含量均較高。提取時(shí)間考察了 0.25、0.5、1.0 h,結(jié) 果顯示 0.5 h 可提取完全。提取體積考察了 15、25、 50 mL,結(jié)果顯示無(wú)明顯差別,故將提取體積定為25 mL。 流動(dòng)相的確定 考察了乙腈 - 水、乙腈 -0.1% 磷 酸 水 溶 液、甲 醇 -0.1% 磷 酸 水 溶 液、甲 醇 - 水 4 個(gè)溶劑系統(tǒng),結(jié)果顯示以乙腈 -0.1% 磷酸水溶液為 流動(dòng)相,所得色譜峰峰形較好,分離效果最佳;同時(shí) 考察了采用 41% 乙 腈 -0.1% 磷 酸 水 溶 液、42% 乙 腈 -0.1% 磷酸水溶液以及本實(shí)驗(yàn)的梯度洗脫,結(jié)果 以 41% 乙腈 -0.1% 磷酸水溶液為流動(dòng)相時(shí)化合物Ⅲ 出峰時(shí)間太晚,以 42% 乙腈 -0.1% 磷酸水溶液為流 動(dòng)相時(shí)化合物Ⅰ將雜質(zhì)峰包進(jìn)去,故采用本實(shí)驗(yàn)的梯 度洗脫。本實(shí)驗(yàn)對(duì) 6 批不同產(chǎn)地沒(méi)藥藥材中化合 物Ⅰ、Ⅱ和Ⅲ 3 個(gè)成分的含量進(jìn)行測(cè)定,結(jié)果顯示不 同樣品含量差異較大。本文建立了沒(méi)藥中 3 個(gè)主要 倍半萜成分的 HPLC 含量測(cè)定方法,色譜峰分離效果 較好,基線較平,重現(xiàn)性較好,簡(jiǎn)便可靠,可為進(jìn)一步 完善沒(méi)藥藥材的質(zhì)量評(píng)價(jià)方法提供參考依據(jù)。
產(chǎn)品中心 | 技術(shù)支持 | 下載中心 | 在線留言 | 合作伙伴 | 新聞動(dòng)態(tài) | 關(guān)于舒美 | 聯(lián)系我們 |

Copyright © 2002-2020 上海蟻霖科學(xué)儀器有限公司 版權(quán)所有
備案號(hào): 滬ICP備19002068號(hào)
服務(wù)熱線:13621695486
售后電話:13621695486
公司郵箱:yilinkexue@163.com
公司地址:上海浦東新區(qū)金高路2131弄17號(hào)401室
 掃一掃進(jìn)入手機(jī)官網(wǎng)
掃一掃進(jìn)入手機(jī)官網(wǎng)
 掃一掃關(guān)注我們
掃一掃關(guān)注我們
